generalitet
Cystisk fibrose er den mest almindelige autosomale recessive sygdom i den kaukasiske befolkning, der påvirker ca. 1 person i hver 2.500.
Denne patologiske tilstand er kendt for dets skadelige virkninger på åndedrætssystemet, men det påvirker også andre systemer som fordøjelses- og reproduktive systemer.
Hos personer med cystisk fibrose er luftvejene tilstoppede med tykt, viskøst slim, hvilket er vanskeligt at fjerne selv med den mest energiske hoste. Åndedræt bliver svært, og patienter - hvis der ikke gøres en kontinuerlig indsats for at holde luftvejene rene flere gange om dagen - risikerer de at dø på grund af deres egen udskillelse. Cystisk fibrose patienter dør ofte på grund af lungebetændelse, da de tilstoppede luftveje giver et frugtbart miljø for bakteriel vækst.

Årsager
Cystisk fibrose er forårsaget af mutationer i cystisk fibrosis fibrosis (CFTR) transmembran konduktansgen lokaliseret ved kromosom 7 (locus mapping: 7q31).
Mindst 1500 mutationer af CFTR genet er kendte. Den hyppigste mutation kaldes almindeligvis "Delta-F508" (DF508) og bestemmes ved deletionen af 3 basepar i exon 10, hvilket resulterer i tab af phenylalanin i position 508.
Proteinet kodet af CFTR-genet er en transmembrankanal, der tilhører ATPase-superfamilien af menneskehandel eller ABC-transporters, placeret i niveauet af den episke cellemes apikale membran og deputeret til transporten af chlorion.
Under normale forhold udskiller særlige celler, der styrer luftvejene, slim sammen med en vandig væske, som nedsætter deres tæthed. Ved cystisk fibrose nedsættes sekretionen af den vandige væske kraftigt , derfor bliver slimmet meget tæt og vanskeligt at fjerne fra luftvejene.
I luftvejsepitelet, som alle epithelier, der bærer væsker, afhænger vandtransport af transport af opløste stoffer. For at udskille vand transporterer cellerne i respiratorisk epitel aktivt klorioner (Cl-) fra interstitielvæsken til lumenet, hvilket skaber et negativt elektrisk potentiale, der forårsager et passivt strøm af natrium (Na +) i samme retning. Bevægelserne af Na + og Cl- hæver væskens osmotiske tryk, der vækker epithelens hældning vendt mod lumen, hvorfor vandet bevæger sig passivt i overensstemmelse med den osmotiske gradient, fra den interstitielle væske til lumenet. Gendefekten, der påvirker forekomsten af cystisk fibrose, forhindrer direkte transporten af Cl- og indirekte forstyrrer transporten af Na + og vand. Følgelig er den osmotiske gradient, der er nødvendig for udskillelsen af vand, ikke skabt i epithelet.
Risikofaktorer
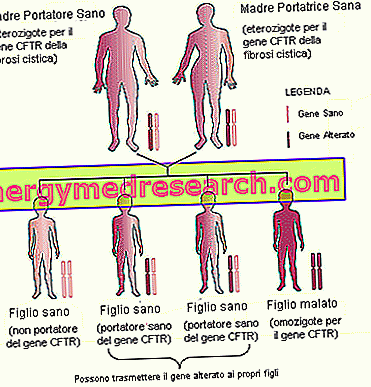
- Familie arv . I betragtning af at cystisk fibrose er en arvelig sygdom, som overføres på autosomal recessiv måde, er det vigtigt at overveje fremtidens forældres familiehistorie (anamnese).
- Race . Forekomsten af cystisk fibrose er større hos hvide mennesker i nord og europæisk oprindelse.

Symptomer og kliniske tegn
For at lære mere: Cystiske fibrose Symptomer
Sværhedsgraden af symptomer kan variere afhængigt af sygdomsforløbet: De fleste kliniske tegn påvirker åndedrætssystemet og mavetarmsystemet.
Respiratoriske tegn og symptomer
Det tykke og viskose slim i forbindelse med cystisk fibrose forårsager luftvejsobstruktion og hæmmer aktiviteten af cilia, der linjer lungepitelet, hvilket letter elimineringen af selve slimhinden. I de små luftveje bidrager den resulterende slimhinde stagnation til at skabe et ideelt miljø for bakterier, såsom Streptococcus aureus eller Pseudomonas aeruginosa, som prædisponerer det berørte individ for tilbagevendende infektioner og betændelser, der igen kan være svære, lignende lungebetændelse. Den kroniske karakter af disse inflammatoriske processer kan beskadige lungevæv over tid og følgelig føre til dårlig åndedrætsfunktion.
De karakteristiske symptomer, som udvikles i løbet af cystisk fibrose, omfatter således:
- Vedvarende hoste med sputumproduktion
- Wheezing breath
- Åndenød og vejrtrækningsbesvær
- Tilbagevendende lungeinfektioner
Fordøjelsessystemet tegn og symptomer
Tykt slim kan også lukke kanalerne, der bærer fordøjelsesenzymer fra bukspyttkjertlen til tyndtarmen. Uden disse fordøjelsesenzymer kan tarmene ikke fordøjes og absorberer næringsstoffer fuldstændigt i den taget mad (især fedtstoffer, kolesterol og fedtopløselige vitaminer A, D, E og K).
Resultatet er ofte følgende:
- Hævet abdomen, meteorisme, steatorrhea
- Dårlig vægtforøgelse og dårlig vækst (på grund af underernæring)
- Intestinal blokering, især hos nyfødte (meconium ileum)
- Forstoppelse eller alvorlig forstoppelse
Komplikationer
Komplikationer i åndedrætssystemet
- Bronchiectasia : Cystisk fibrose er en af hovedårsagerne til bronchiectasia, en tilstand, der beskadiger luftveje, hvilket resulterer i obstruktiv ventilatorfejl, med en markant reduktion i udåndingsstrømmen.
- Kroniske infektioner : Det tykke slim, som stagnerer i lungerne og bihulerne, giver en ideel yngleplads til bakterier og svampe. Som følge heraf kan personer med cystisk fibrose have hyppige angreb af bihulebetændelse, bronkitis eller lungebetændelse.
- Næsepolypper : Siden næsens indre beklædning er betændt og opsvulmet, kan der udvikles bløde kødfulde vækst (polypper), som kan forhindre vejrtrækning under søvn.
- Hoste med blod (hæmoptyse): Over tid kan cystisk fibrose forårsage udtynding af luftvejsvæggene. Som følge heraf kan patienter med cystisk fibrose udlede blod fra luftvejene, normalt gennem hoste.
- Pneumothorax : Denne tilstand, der består af ophobning af luft i pleurhulen, er mere almindelig hos ældre med cystisk fibrose. Pneumothorax kan forårsage brystsmerter og åndenød.
- Lungekollaps : Kroniske lungeinfektioner kan skade lungeparenchyma, hvilket gør lungekollaps mere sandsynligt.
- Respiratorisk insufficiens : Over tid kan cystisk fibrose beskadige lungevæv og kompromittere dets funktionalitet.
Komplikationer af fordøjelsessystemet
- Ernæringsmæssige mangler : Tykt slim kan blokere de bugspytkirtelkanaler, der bærer fordøjelsesenzymer fra bugspytkirtlen til tyndtarmen. Uden disse enzymer kan kroppen ikke absorbere proteiner, fedtstoffer og fedtopløselige vitaminer.
- Diabetes : Pancreas-beta-celler udskiller insulin, hvilket er nødvendigt for at medlede blodglukose regulering. Cystisk fibrose øger risikoen for udvikling af diabetes. Ca. 20 procent af de ramte personer udvikler diabetes i alderen 30, så patienterne testes regelmæssigt for blodglukose.
- Forhindring af galdekanalerne : Kanalen, der bærer galde fra leveren, og fra galdeblæren til tyndtarmen kan blive blokeret og betændt, hvilket fører til leverproblemer og nogle gange galdesten.
- Rektal prolaps : hyppig hoste eller anstrengelse under afføring (forstoppelse) kan forårsage, at det indre rektalvæv stikker ud af anus.
- Intussusception : børn med cystisk fibrose har større risiko for intussusception, en tilstand, hvor en del af tarmfoldene foldes tilbage på sig selv og skaber invaginationer. Resultatet er intestinal obstruktion, en nødsituation, der kræver øjeblikkelig behandling.
Komplikationer af det reproduktive system
Næsten alle mænd med cystisk fibrose er sterile, fordi kanalerne, som forbinder testiklerne og prostatakirtlen (vas deferens) er blokeret af slim eller mangler helt (fraværet af kanalerne er stadig en årsag ukendt).
Selv om kvinder med cystisk fibrose kan være mindre frugtbare end andre kvinder, beholder de stadig evnen til at blive gravid og bringe graviditeten til ophør. Imidlertid kan svangerskabet bidrage til en forværring af symptomer på cystisk fibrose.
Andre komplikationer
Osteoporose : personer med cystisk fibrose har en højere risiko for at udvikle osteoporose med deraf følgende tab af masse og knoglestyrke. Patienter bør rådes til at tage calcium, D-vitamin og bisfosfonater efter behov.
Elektrolyt ubalancer : personer med cystisk fibrose har en tendens til at have et højere end normalt niveau af salt i sved. Tegn og symptomer som øget hjertefrekvens, træthed, svaghed og lavt blodtryk hjælper med at fremhæve den dekompensation, som hydro-saltopløsningen i blodet gennemgår.
Diagnose og terapi af cystisk fibrose »


