Hvad er Marfan Syndrome?
Marfan syndrom beskriver en kompleks arvelig sygdom i bindevævet, som primært påvirker øjnene, kardiovaskulærsystemet og skeletmuskelsystemet. Men i betragtning af at hvert organ består af bindevæv, kan Marfan syndrom ideelt set ødelægge og forstyrre stærkt med vækst og funktion af hvert anatomisk sted.
Syndromet overføres som et autosomalt dominerende træk: vi står derfor over for en alvorlig genetisk sygdom, der har en ekstremt variabel fænotypisk ekspression (mangler kan variere enormt fra familie til familie eller fra patient til patient).
Hvad udløser Marfan syndrom er ændringen af FBN1 genet (på kromosom 15), som koder for fibrillin-1, et meget vigtigt bindende glycoprotein, der udgør den strukturelle understøttelse af mikrofibriller.
Mikrofibriller: bestående af fibrillin, mikrofibrillerne er til stede i den ekstracellulære matrix, hvori de danner en bane til aflejring af elastinet i de elastiske fibre. Selvom allestedsnærværende i kroppen, findes mikrofibriller hovedsagelig i aorta, ledbånd og zonules af ciliære legemer (på okularniveau).
At være en autosomal dominerende sygdom er kun børn, der har arvet et FBN-1-gen, der ændres af begge forældre, påvirket af Marfan syndrom. Ikke desto mindre er sygdommen i et tilfælde ud af 4 resultatet af spontane mutationer hos patienter, der ikke har familiehistorie.
Navnet på sygdommen kommer fra den franske børnelæge, der først beskrev det i 1896 (A. Marfan), hvorefter det måtte vente til 1991 for at identificere det ændrede gen involveret i symptomatisk manifestation: opdageren var F. Ramirez.
Se videoen
X Se videoen på youtubeÅrsager
Vi har nævnt, at Marfan syndrom er den umiddelbare udtryk for mutationen af et gen, som koder for fibrillin-1. Lad os forsøge at uddybe emnet for at forstå, hvilken mekanisme der er etableret for at udløse syndromet.
FIBRILLINA 1 er en glycoproteinkomponent af elastin, der er afgørende for at sikre og opretholde elasticitet og vævsstyrke. I fysiologiske tilstande binder fibrillin 1 til et andet protein, kendt som TGF-beta (eller transformerende vækst-beta-faktor). TGF-beta ser ud til at være involveret i skadelige processer, der påvirker vaskulær glatmuskel og den ekstracellulære matrix. Ud fra disse antagelser er nogle forfattere overbeviste om, at Marfan syndromet ud over mutationen af FBN-1 genet også skyldes et overskud af TGF-beta, især i aorta, i hjerteventiler og i lungerne.
incidens
Det anslås, at Marfan syndrom påvirker 1 individ hver 3.000-5.000 fødsler og manifesterer sig usædvanligt mellem hanner og kvinder, uden forkærlighed til en præcis race. Statistikker viser, at 75% af patienterne har en positiv familiehistorie; i de resterende 25% ligger årsagen i sporadiske mutationer, der tilsyneladende er forbundet med en eller anden måde med faderens avancerede alder på tidspunktet for opfattelsen.
Børn med ekstremt alvorlige former for Marfan syndrom har en forventet levetid på mindre end et år.
Før udviklingen af åben hjerte kirurgiske strategier havde de fleste patienter med Marfan syndrom en gennemsnitlig levetid på 32 år; Takket være den konstante forbedring af medicinske og farmakologiske terapier lever i øjeblikket Marfan syndrom patienter i gennemsnit op til 60 år.
Tegn og symptomer
For at lære mere: Symptomer Marfan syndrom
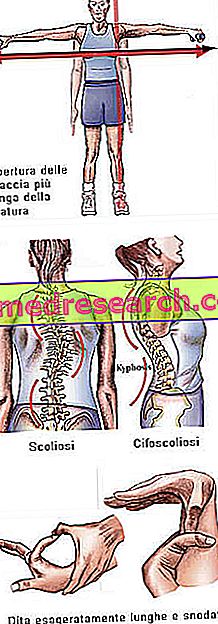
Marfan syndrom kan køre helt asymptomatisk. Berørte patienter har en for slank struktur, der er uforholdsmæssigt høje og tynde. De nedre og øverste lemmer har en meget højere længde end stammen (dolicostenomegalia). Der er også tale om araknodactyly at udtrykke det bedste udtryk for fingers overdrevne længde, typisk for emner, der er berørt af Marfan syndrom: Hænderne er derfor sammenlignet med en edderkoppers ben.
Med hensyn til højde har disse patienter en gennemsnitlig højde over det 97. percentil.
Andre særpræg, der ofte findes hos patienter med Marfan syndrom, omfatter:
- Arme åbne større end højde
- Løse ledd → Overdreven fælles bevægelighed
- Deformation af brystvæggen
- Krystallinsk fortrængning
- Øverste del af kroppen mindre udviklet end den nederste del
- Spontan pneumothorax (11%)
- skoliose
- Hudstriae på lår, ryg, deltoid, pectoral niveau
Blandt de mest problematiske tegn, der er forbundet med Marfan syndrom, nævner vi hjerteventil-prolaps og mitralventilinsufficiens: En lignende tilstand kan let fremme dilation af aorta-ring og aorta-dissektion.
Tabellen viser de tegn, der kan findes hos patienter med Marfan syndrom. Tegnene der er beskrevet der er ikke altid til stede, men en god del af disse findes.
| Berørt anatomisk sted | Mulige symptomer |
Cute | Striae i thoracic, lumbal og sacral area |
Øjne | Nedsat syn, astigmatisme, retinal løsrivelse, snævert vinkelglaukom, linsforskydning, myopi |
Benstruktur | Artralgi, kyphoscoliosis, dolicostenomelia (overdreven udvikling i lem længde med hensyn til stammen), hypermobilitet, høj gane, deformeret bryst, flade fødder, smalle og tynde håndled, unormal tilbagekaldelse / brystproteser, skoliose, buede skuldre, spondylolistese |
Dita | arachnodactyly |
Lunger | Spontan pneumothorax, dyspnø, idiopatisk lungeobstruktiv sygdom |
Ansigtsændringer | Ogival gane (misdannelse af ganen), mandibular retrognathia (defekt i kæbenudvikling), langstrakt ansigt |
hjerte | Angina pectoris, abdominal aorta aneurisme, hjertearytmi, thorax aorta dilation / brud / dissektion, aorta insufficiens, mitral ventil prolapse |
sprog | Sprogproblemer |
diagnose
I betragtning af de mere end 200 mulige mutationer er anvendelsen af genetiske markører næsten umulig til diagnostiske formål.
Opgørelsen af Marfan syndrom er ikke altid så øjeblikkelig, idet fænotypisk ekspression af mutationen ikke altid er indlysende og let at identificere. Den diagnostiske forsinkelse kan alvorligt kompromittere patientens overlevelse: tænk f.eks. På manglende anerkendelse af et kardiovaskulært problem.
Diagnostiske kriterier for Marfan syndrom blev udarbejdet internationalt i 1996: diagnosen består i undersøgelsen af familiens historie forbundet med en kombination af store og mindre indikatorer for syndromet.
Nogle af de mange anvendte diagnostiske tests er:
- ekkokardiografi
- Magnetisk angiorison og CT (til undersøgelse af aorta)
- magnetisk resonans angiografi (MRA) med kontrastvæske (for at gøre interne strukturer af aorta tydelige)
- undersøgelse med glidelamper (for at analysere den mulige dislokation af linsen)
- måling af okulært tryk (for at fremhæve den mulige tilstedeværelse af glaukom)
- genetiske tests (anbefalet før man opdager et barn for at fastslå syndromet eller ej)
behandlingsformer
At være en genetisk sygdom, der er ingen medicin eller behandling, der kan reversere sygdommen.
Brug af medicin er imidlertid afgørende for at mildne symptomerne og undgå eventuelle komplikationer, især hjertesygdomme. Til dette formål er lægemidler til reduktion af blodtrykket, såsom sartans (især), ACE-hæmmere og beta-blokkere, særligt egnede.
I forbindelse med Marfan syndrom kan patienter, der lider af skoliose, følge særlig pleje såvel som for dem, der er ramt af glaukom.
Kirurgi er tænkelig at korrigere den anomale aorta dilatation, et element der ofte forener flertallet af patienter med Marfan syndrom.
Fortsæt: Marfans syndrom - narkotika og pleje »